Jak powstaje lek?

Etapy powstawania nowego leku
- Badania - nowy lek tworzy się w oparciu o potrzeby pacjentów chorych na określone schorzenie oraz szeroką i najbardziej aktualną wiedzę na temat danej jednostki chorobowej.
- Cel - w oparciu o tę wiedzę tworzy się molekułę (cząsteczkę) lub wiele cząsteczek o podobnej strukturze.
- Cząsteczka - uzyskuje się cząsteczkę wiodącą o najbardziej obiecujących właściwościach.
- Optymalizacja - w warunkach laboratoryjnych sprawdza się, czy wybrana molekuła (która jeszcze nie jest lekiem) wywołuje pożądane działanie biologiczne.
- Faza laboratoryjna - jeżeli uda się dowieść, że nowa molekuła ma przewidywane działanie, rozpoczyna się etap badań na zwierzętach, który pozwala sprawdzić potencjalne działania toksyczne. Takie badanie może trwać nawet kilka lat.
Start badań nowego leku - badania kliniczne (fazy)
Uzyskanie zgody na prowadzenie badania
Aby rozpocząć badania z udziałem pacjentów, konieczne jest złożenie wniosku o wydanie pozwolenia na prowadzenie badania klinicznego oraz uzyskanie zgody Urzędu Rejestracji oraz Komisji Etycznej. W Polsce wnioski dotyczące możliwości przeprowadzenia badania składa się do:
- Prezesa Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych oraz
- komisji bioetycznej.
Proces uzyskiwania opinii właściwej komisji bioetycznej i pozwolenia Prezesa URPL mogą być prowadzone równolegle.
Prezes Urzędu dokonuje oceny takiego wniosku w terminie nie dłuższym niż 60 dni. Termin ten liczy się od dnia złożenia pełnej dokumentacji badania klinicznego. W trakcie postępowania o wydanie pozwolenia na prowadzenie badania klinicznego, Prezes Urzędu może jednorazowo żądać od sponsora Sponsor dostarczenia informacji uzupełniających, niezbędnych do wydania pozwolenia. Włączenie pacjenta do badania przed uzyskaniem pozwolenia Prezesa Urzędu nie jest możliwe.
Komisja bioetyczna wydaje opinię o badaniu klinicznym podczas posiedzenia uwzględniając kryteria etyczne, celowość i wykonalność badania.
Zgodnie z przepisami sponsor Sponsor może rozpocząć badanie kliniczne Badanie kliniczne dopiero po uzyskaniu zarówno zgody Prezesa Urzędu, jak i pozytywnej opinii komisji bioetycznej.
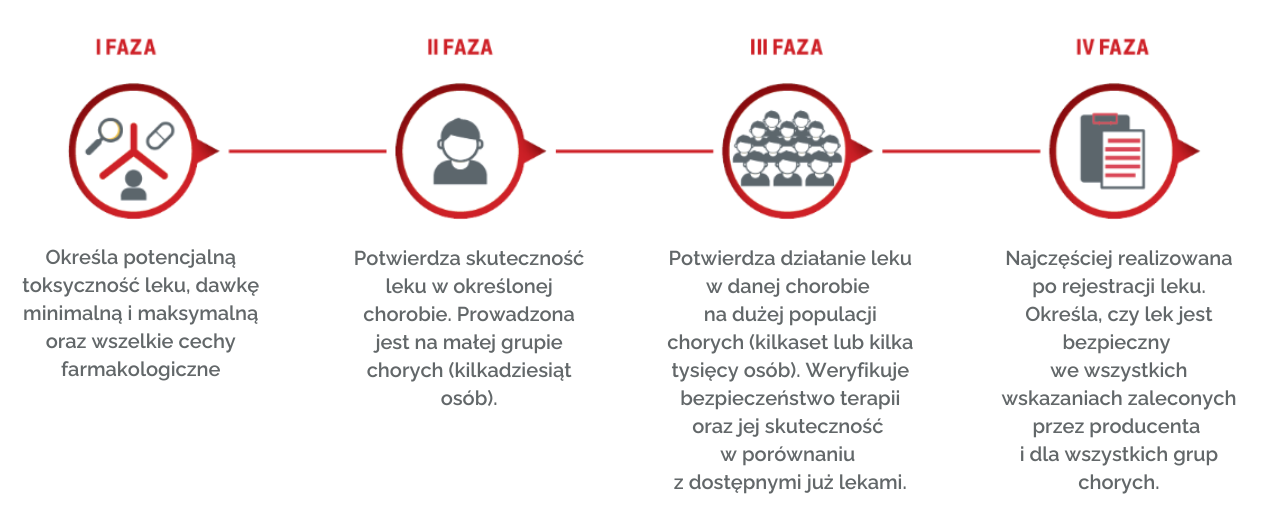
Fazy badań z udziałem pacjentów:
- I faza - określa potencjalną toksyczność leku, dawkę minimalną i maksymalną oraz wszelkie cechy farmakologiczne przyszłego leku (np. jak substancja wydala się z organizmu, jak długo działa). Badania tej fazy prowadzone są wyłącznie na ochotnikach.
- II faza - potwierdzenie skuteczności i bezpieczeństwa leku w danej chorobie. Ta część badań prowadzona jest na małej grupie chorych (kilkadziesiąt osób).
- III faza - badacze Badacz potwierdzają działanie leku w danym wskazaniu na dużej populacji chorych (kilkaset lub nawet kilka tysięcy osób), jednocześnie sprawdzane jest bezpieczeństwo danej terapii. Weryfikuje się także, czy dany lek jest skuteczniejszy od dotychczas stosowanego standardu.
- IV faza - prowadzone są po dopuszczeniu produktu leczniczego Produkt leczniczy do obrotu. Etap ten ma określić, czy lek jest bezpieczny we wszystkich wskazaniach zalecanych przez producenta i dla wszystkich grup chorych. Prowadzona jest też weryfikacja wyników uzyskanych w poprzednich etapach.
Rejestracja nowego leku
Pacjent może kupić lek w aptece dopiero wtedy, gdy właściwy organ wyda decyzję o pozwoleniu na dopuszczenie do obrotu. Przed wydaniem decyzji właściwy organ potwierdza, czy dany produkt leczniczy Produkt leczniczy jest odpowiedniej jakości, czy jest bezpieczny. Ocena odbywa się na podstawie dokumentacji złożonej przez podmiot odpowiedzialny.
- W Polsce organem właściwym jest Prezes Urzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. Prezes Urzędu wydaje decyzję w sprawie dopuszczenia do obrotu produktów leczniczych, rejestrowanych na drodze procedury narodowej, wzajemnego uznania (MRP) i zdecentralizowanej (DCP). W rezultacie procedury narodowej pozwolenie na lek jest udzielane tylko na terytorium Polski, zaś w przypadku procedur MRP i DCP, zwanych europejskimi, decyzję o udzieleniu pozwolenia na dopuszczenie do obrotu wydaje się w krajach UE równocześnie biorących udział w procedurze.
- Komisja Europejska wydaje pozwolenie na dopuszczenie do obrotu dla produktów leczniczych Produkt leczniczy rejestrowanych na drodze procedury scentralizowanej (CP). Dokumentacja rejestracyjna w tej procedurze jest oceniana przez Europejską Agencję Leków. W rezultacie procedury scentralizowanej wydawane jest jedno pozwolenia na dopuszczenie do obrotu, które pozwala na wprowadzanie do obrotu leku we wszystkich państwach członkowskich UE.
- Po wydaniu decyzji rejestracyjnej lek może zostać wprowadzony do obrotu.
- O tym, czy nowy lek będzie refundowany w konkretnym wskazaniu, decyduje w Polsce Ministerstwo Zdrowia. Aby tak się stało, właściciel pozwolenia na dopuszczenie do obrotu leku musi złożyć dodatkową dokumentację, a Ministerstwo występuje o rekomendację do Agencji Oceny Technologii Medycznych i Taryfikacji (AOTMiT). Agencja dokonuje analiz m.in. na podstawie artykułów naukowych, opinii ekspertów czy Rady Przejrzystości i wydaje rekomendację, czy warto czy też nie, refundować dany lek. Rekomendacja Agencji nie jest wiążąca dla Ministerstwa.