Rozporządzenie UE w sprawie wyrobów medycznych (MDR)
Rozporządzenie UE w sprawie wyrobów medycznych Wyrób medyczny 2017/745 („rozporządzenie MDR”) weszło w życie 26 maja 2021 r. i zastępuje dotychczas obowiązującą dyrektywę Rady Europejskiej 93/42/EWG (tzw. „dyrektywę MDD”). Zgodność z rozporządzeniem MDR jest obowiązkowa dla przedsiębiorców, decydujących się na sprzedaż wyrobów medycznych na rynku europejskim.
Nowe przepisy zakładają ustanowienie wysokich norm jakości i bezpieczeństwa dla wyrobów medycznych oraz harmonizację zasad wprowadzania ich do obrotu i używania na rynku unijnym. Ponadto proponowane rozwiązania mają zagwarantować szerszą identyfikację wyrobów medycznych i ich producentów, a także innych podmiotów w łańcuchu dostaw, zapewniając tym samym skuteczniejszy nadzór, a użytkownikom łatwiejszy dostęp do informacji nt. wyrobów.
Najważniejsze zmiany wprowadzone rozporządzeniem:
A. NOWE REGUŁY KLASYFIKACJI
Rozporządzenie MDR określa nowe reguły klasyfikacji wyrobów medycznych. W zależności od czasu stosowania, stopnia inwazyjności lub możliwości ponownego wykorzystania wyróżnia się wyroby medyczne Wyrób medyczny klasy I, IIa, IIb oraz III. Klasyfikacja wyrobu ustalana jest przez producenta. Co istotne, zgodnie z nowymi wytycznymi część wyrobów wymaga przeklasyfikowania do wyższych klas.
B. OCENA ZGODNOŚCI
Dla każdego wyrobu medycznego Wyrób medyczny przed wprowadzeniem wyrobu do obrotu należy przeprowadzić tzw. „ocenę zgodności”. Ważne jest, że wszystkie wyroby klasy wyższej niż I wymagają udziału jednostki notyfikowanej w ocenie zgodności. W przypadku wyrobów klasy I procedurę oceny zgodności przeprowadza samodzielnie producent. Wyjątek stanowią wyroby klasy I, które są wprowadzane do obrotu w stanie sterylnym, posiadają funkcję pomiarową lub są narzędziami chirurgicznymi, w tym przypadku udział jednostki w ocenie zgodności jest ograniczony do tych poszczególnych aspektów. Wyroby medyczne po przeprowadzeniu właściwej według klasy oceny zgodności należy oznakować znakiem CE.
C. PODMIOTY W ŁAŃCUCHU DOSTAW WYROBÓW MEDYCZNYCH
Rozporządzenie MDR definiuje podmioty gospodarcze obecne w łańcuchu dostaw w następujący sposób:
- producent oznacza osobę fizyczną lub prawną, która wytwarza lub całkowicie odtwarza wyrób lub która zleca zaprojektowanie, wytworzenie lub całkowite odtworzenie wyrobu i oferuje ten wyrób pod własnym imieniem i nazwiskiem lub nazwą lub znakiem towarowym;
NOWOŚĆ! Pojęcie „producenta” zastępuje dotychczas używaną w dyrektywie MDD nomenklaturę tj. „wytwórcy” wyrobów medycznych.
- upoważniony przedstawiciel oznacza osobę fizyczną lub prawną mającą miejsce zamieszkania lub siedzibę w Unii, która otrzymała i przyjęła od producenta, który ma siedzibę poza Unią, pisemne upoważnienie do występowania w imieniu producenta w zakresie określonych zadań w odniesieniu do obowiązków producenta wynikających z Rozporządzenia MDR (jeśli producent nie ma siedziby na terytorium UE, powinien wyznaczyć swojego upoważnionego przedstawiciela);
- dystrybutor oznacza osobę fizyczną lub prawną w łańcuchu dostaw, inną niż producent lub importer, która udostępnia wyrób na rynku, do momentu wprowadzenia do używania (jest to bardzo szeroka grupa pomiotów, do której można zaliczyć przykładowo hurtownie farmaceutyczne, apteki, czy też sklepy medyczne);
- importer oznacza osobę fizyczną lub prawną, mającą miejsce zamieszkania lub siedzibę w Unii, która wprowadza do obrotu w Unii wyrób z państwa trzeciego.
D. PRZYKŁADOWE OBOWIĄZKI PODMIOTÓW GOSPODARCZYCH WG. ROZPORZĄDZENIA MDR
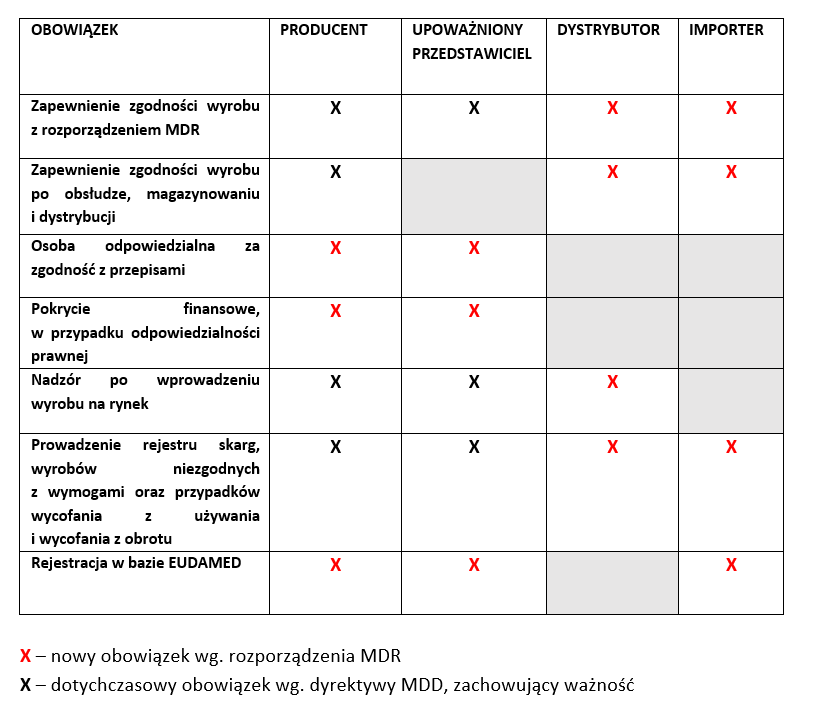
E. OBOWIĄZKOWE WDROŻENIE SYSTEMU ZARZĄDZANIA JAKOŚCIĄ PRZEZ PRODUCENTA
System zarządzania jakością (SZJ) obejmuje wszystkie części i elementy organizacji producenta zajmujące się jakością procesów, procedur i wyrobów. Zarządza on nie tylko strukturą i obowiązkami, ale również procedurami, procesami i zasobami w zakresie zarządzania niezbędnymi do tego, by wdrożyć zasady i działania potrzebne do osiągnięcia zgodności z przepisami rozporządzenia MDR. Wdrożony u producentów SZJ powinien odpowiadać wymogom normy zharmonizowanej ISO 13485. Wśród aspektów objętych SZJ powinno wyróżniać się m.in. zarządzanie ryzykiem, zarządzanie działaniami korygującymi i zapobiegawczymi oraz weryfikację ich skuteczności, odpowiedzialność kadry zarządzającej, a także zarządzanie zasobami, w tym wybór i kontrolę dostawców i podwykonawców.
System zarządzania ryzykiem stanowi jeden z integralnych elementów SZJ. Producenci, prowadząc system zarządzania ryzykiem m.in. ustanawiają i dokumentują plan zarządzania ryzykiem dla każdego wyrobu; identyfikują i analizują znane i przewidywane zagrożenia związane z każdym wyrobem oraz oszacowują i oceniają ryzyko związane z przewidzianym używaniem oraz ryzyko występujące podczas przewidzianego używania i racjonalnie przewidywalnego nieprawidłowego użycia danego wyrobu. Sprawozdanie z wdrożonej analizy ryzyka powinno być przeprowadzone zgodnie z normą ISO PN-EN 14971.
F. PRZECHOWYWANIE DOKUMENTACJI
Rozporządzenie MDR nakłada na producentów obowiązek sporządzania i bieżącej aktualizacji dokumentacji technicznej dotyczącej wyrobów medycznych, innych niż wykonanych na zamówienie.
Co istotne, dokumentacja techniczna powinna być sporządzona w sposób jasny, uporządkowany i jednoznaczny, umożliwiający łatwe wyszukiwanie poszczególnych elementów obejmujących:
- opis i specyfikację wyrobu,
- wzmiankę o poprzednich i podobnych generacjach wyrobu,
- zestaw etykiet oraz instrukcji używania,
- informacje o projekcie i produkcji wyrobu,
- informacje dotyczące potwierdzenia zgodności z procedurami dotyczącymi bezpieczeństwa i działania,
- analizę stosunku korzyści do ryzyka wynikającego z używania wyrobu, a także informacje o rozwiązaniach przyjętych w celu zarządzania ryzykiem,
- wyniki wszystkich testów lub badań weryfikacyjnych i walidacyjnych wraz z ich krytyczną analizą, w tym dane przedkliniczne i kliniczne,
- dokumentację techniczną dotyczącą nadzoru po wprowadzeniu wyrobu do obrotu.
Producenci i upoważnieni przedstawiciele zobowiązani są do przechowywania – do dyspozycji URPL– dokumentacji technicznej, deklaracji zgodności UE oraz, w stosowanych przypadkach, kopii odpowiednich certyfikatów, przez okres co najmniej 10 lat od wprowadzenia do obrotu ostatniego wyrobu objętego deklaracją zgodności UE. W przypadku wyrobów do implantacji okres wynosi co najmniej 15 lat po wprowadzeniu do obrotu ostatniego wyrobu.
G. ZMIANY W ZAKRESIE BADAŃ KLINICZNYCH
Problematyka badań klinicznych została szczegółowo uregulowana w rozdziale VI rozporządzenia MDR. Nowe przepisy znacząco zaostrzają dotychczasowo obowiązujące regulacje prawne. Rozporządzenie MDR doprecyzowuje bowiem, w jaki sposób projektuje się, dopuszcza, prowadzi, rejestruje te badania, a także powiadamia oraz sprawozdaje w ich zakresie. Doregulowano także rozwiązania mające zastosowanie przy przeprowadzaniu oceny klinicznej, której przeprowadzenie jest obowiązkiem każdego producenta wyrobów medycznych – bez względu na klasę wyrobu. Ocena kliniczna umożliwia określenie poziomu akceptowalności stosunku korzyści do ryzyka w warunkach użytkowania wyrobu medycznego, będącego elementem systemu zarządzania jakością.
H. KOD INDENTYFIKACYJNY WYROBU (UDI) I BAZA EUDAMED
Zgodnie z rozporządzeniem MDR wszystkie wyroby medyczne będą musiały być wyposażone w unikalny kod identyfikacyjny wyrobu (ang. Unique Device Identification, UDI), umieszczony na etykiecie i opakowaniu oraz w przypadku niektórych wyrobów, na samym produkcie. Znakowanie produktów kodem UDI, pozwoli na zwiększenie możliwości śledzenia wyrobów medycznych w całym łańcuchu dostaw przez połączenie wszystkich informacji na temat poszczególnych wyrobów w cyfrowej bazie informacji o nazwie EUDAMED. Nośnik UDI powinien być przymocowany bezpośrednio do wyrobu medycznego lub jego opakowania.
I. NOWE OBOWIĄZKI INSTYTUCJI ZDROWIA PUBLICZNEGO
„Instytucja zdrowia publicznego” to organizacja, której podstawowym celem jest opieka nad pacjentami lub leczenie pacjentów lub promowanie zdrowia publicznego. Jako instytucje zdrowia publicznego będą kwalifikowane szpitale i inne podmioty lecznicze. Do obowiązków instytucji zdrowia publicznego będzie należało:
- zachowanie i przechowanie – najlepiej w formie elektronicznej – kodów UDI wyrobów, które dostarczyły lub które zostały im dostarczone, jeżeli wyroby te należą do wyrobów do implantacji klasy III.
- zgłaszanie incydentów dotyczących wyrobów medycznych
Producent będzie zobowiązany dostarczyć do każdego wyrobu medycznego do implantacji – „kartę implantu”, zawierającą wszelkie istotne informacje nt. wyrobu. Natomiast każda instytucja zdrowia publicznego musi udostępnić pacjentowi, któremu wszczepiono dany wyrób wraz z kartą implantu szczegóły produktu, takie jak m.in. dane identyfikacyjne wyrobu, ostrzeżenia czy środki ostrożności.